桑迪亚国家实验室开发的LAMMPS软件主要用于模拟什么?
参考资料

参考资料


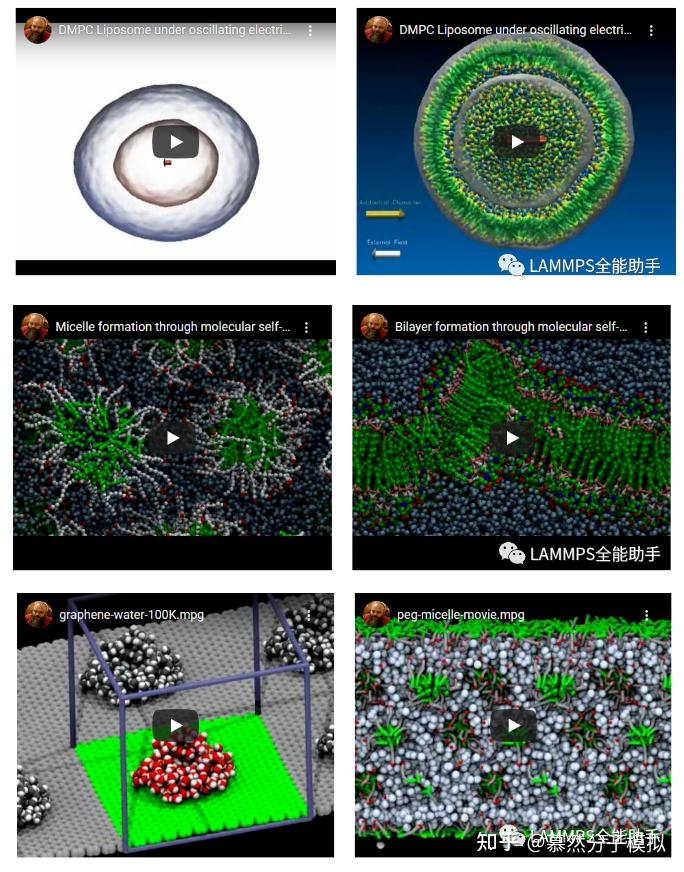

Moonshot回答
LAMMPS软件是由桑迪亚国家实验室(Sandia National Laboratories)开发的分子动力学模拟工具。它主要用于模拟液态、固态或气态的粒子的系综,包括但不限于:
- 研究金属材料的力学性能,例如模拟金属材料的塑性变形和断裂行为。
- 研究高分子材料的分子构象。
- 模拟全原子、聚合物、生物、固态(金属、陶瓷,氧化物)、粒状和粗料化体系。
- 计算微观结构的演化,例如观察粒子在合理力场作用下的行为。
- 进行结构优化、过渡态搜索和热导率计算等。
LAMMPS软件以其强大的并行计算能力和广泛的应用领域,在材料、物理、化学等模拟领域中发挥着重要作用。它使用MPI实现多机器并行计算,并在新版本中支持基于CUDA和OpenCL的GPU计算。LAMMPS以GNU通用公共许可证发布,因此是开源且免费的。更多信息可以参考LAMMPS Wikiwand和桑迪亚国家实验室维基百科。