参考资料

Moonshot回答
分子力学模拟常用的软件包括:
-
LAMMPS:由美国Sandia国家实验室开发,是一个开源的大规模原子/分子并行模拟器。它支持多种势能函数,适用于软物质、固体力学、材料科学等领域。LAMMPS支持多种并行计算方式,包括MPI和GPU加速。
-
AMBER:是一个广泛使用的分子动力学模拟软件包,特别适用于生物分子系统。它提供了一套完整的工具来处理分子动力学模拟、自由能计算等任务。
-
GROMACS:是一个用于研究生物分子体系的分子动力学软件,特别适合于模拟蛋白质、脂质和核酸等生物大分子。
-
CHARMM:是一个分子模拟程序,广泛应用于模拟生物分子体系,如蛋白质、核酸、脂质等。
-
Materials Studio:是一个综合性的材料模拟平台,提供了分子力学、分子动力学、蒙特卡洛模拟等多种模拟方法。
-
NAMD:是一个用于模拟大生物分子体系的并行分子动力学代码,特别适合于长时间、大规模的模拟任务。
-
OpenMM:是一个用于模拟生物分子体系的开源软件,支持多种力场,并且可以与Python等编程语言集成。
-
MindSpore SPONGE:由华为云社区开发,是一个新一代分子模拟库,具有完全自主的知识产权,使用模块化设计,支持新的力场和抽样方法。
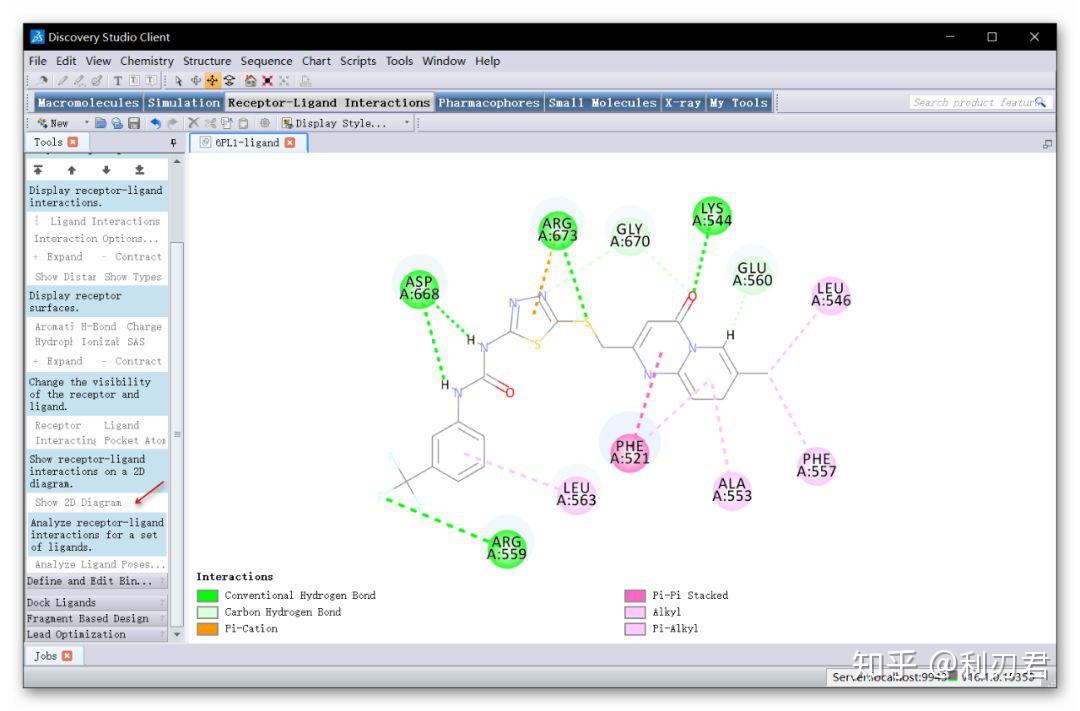
这些软件各有特点,适用于不同的研究领域和应用场景。例如,LAMMPS适合于材料科学和工程领域,而AMBER和GROMACS更适合于生物分子的模拟。选择合适的软件需要根据研究目标和系统特性来决定。图片来源:分子动力学模拟软件、应用及优劣。
继续提问
相关问题
蔡斯·F·罗宾逊的著作《伊斯兰文明三十人传》如何通过三十位人物的生平展现伊斯兰文明的前千年历史?
《旺角卡门》获得了哪些主要奖项和提名,其中哪位男演员因其表现赢得了最佳男配角奖?
法国国家自然历史博物馆的植物标本馆保存了多少份植物标本?
python的对象如何动态增加属性
马格里布叛乱是如何从阿尔及利亚的局部冲突演变为整个萨赫勒地区的长期战争的?
美国在19世纪和20世纪初期是如何通过政权更迭来扩大其在拉丁美洲的影响力的?
冈政伟的兴趣爱好有哪些方面?
汉堡的气候特点是什么?
在设置偏移配置时,如何选择合适的参考坐标系,以及为什么目前只支持基坐标系的偏移?
不同细分行业的景气度差异较大,哪些行业在智能制造领域的表现仍然保持较好增长?